Содержание

Так как изменения генов могут быть разнообразными (изучено более 100 вариантов), а также они комбинируются с другими патологиями строения хромосомного аппарата, то проявлена синдрома Марфана отличаются.
Классификация заболевания
Встречаются легкие случаи и крайне тяжелые, с быстрой прогрессией нарушения деятельности сердечно-сосудистой, легочной системы, почек. Промежуточное положение между ними занимает синдром Марфана средней тяжести.
Также существует подразделение этого заболевания на клинические формы по наличию поражения систем организма:
- стертая – патология в одной или двух системах со слабыми отклонениями от нормы;
- выраженная – незначительные нарушения в 3 системах или серьезные в 1;
- тяжелая – прогрессирующее нарушение деятельности 3 систем.
Стабильным вариантом течения считается болезнь при наличии изменений в работе или строении органов, которые не прогрессируют на протяжении более 1 года.
Признаки наличия отклонения у детей и взрослых
Проявления патологии многообразны, и их первые признаки могут быть обнаружены при рождении ребенка или постепенно нарастают во взрослом возрасте.
Наиболее тяжело протекает классическая форма, при которой изменения имеются в самом раннем периоде развития.
К ним относятся такие симптомы:
- длина тела при рождении более 53 см, у взрослых достигает 190 см (у женщин 175 — 180);
- туловище короткое, а конечности длинные с тонкими пальцами, паукообразной формы;
- мышцы и подкожная клетчатка развиты слабо;
- небо высокое по типу арки, нос длинный, прикус нарушен;
- суставы с избыточной подвижностью;
- имеются вывихи шейного отдела, искривление позвоночника;
- грудная клетка воронкообразной формы;
- плоскостопие;
- головка бедренной кости вдавливается вглубь таза.

Наиболее опасными являются не внешние дефекты, придающие пациентам характерный вид, а патология строения сердца и сосудов. Именно эти пороки развития определяют продолжительность жизни больных и занимают ведущее место в клинической картине. Наиболее типичные из них:
 Читайте также:
Читайте также:

- стеноз магистральных сосудов – аорты и легочной артерии;
- отверстия в перегородках сердца;
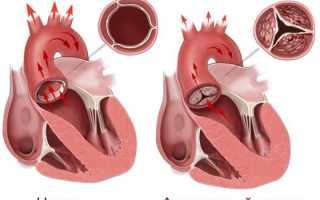
- аортальная недостаточность, расширение восходящего сегмента и аневризмы аорты;
- удлиненные створки митрального клапана с разрывом сухожилий, которые их прикрепляют.

Из-за поражений сосудов и строения клапанного аппарата пациенты страдают от недостаточности кровообращения, они подвержены развитию бактериального эндокардита, аритмии в виде тахикардии, мерцания или трепетания предсердий и желудочков. При комбинированных функциональных и анатомических нарушениях смертельный исход может быть на первом году жизни ребенка.
Нарушения зрения выявляются в виде двустороннего вывиха хрусталика, они, как правило, нарастают до 5 лет и сопровождаются потерей зрения. Также при синдроме Марфана возникают такие офтальмологические патологии:
- близорукость,
- увеличенная и более плоская роговица по сравнению с нормой,
- недоразвитая радужная оболочка,
- косоглазие,
- деформированные сосуды сетчатки.
При нарушении развития твердой оболочки мозга в поясничном отделе позвоночника возникает выпячивание содержимого спинного-мозгового канала, что сопровождается болью и онемением нижних конечностей. В легких возможно формирование эмфиземы, пневмоторакса, почки и мочевой пузырь, а у женщин и матка смещаются книзу, часто происходит растяжение связок, образование паховых и бедренных грыж.
По уровню умственного развития пациенты не отличаются от здоровых людей, а в некоторых случаях имеют очень высокий уровень интеллекта. Это связано с повышенным синтезом адреналина, поэтому такие люди излишне раздражительны, тревожны и возбуждены. Но при этом физическая нагрузка переносится плохо, сопровождается сильной мышечной и головной болью.
Смотрите на видео о синдроме Марфана:
Диагностика заболевания

Основные критерии, по которым можно поставить диагноз (хотя бы один из них):
- расширение или расслоение аорты,
- вывих хрусталика,
- грудная клетка с выпячиванием грудины,
- конечности, длиннее, чем в норме,
- ограничение разгибания локтевых суставов или деформация бедренных.
Если пациент сгибает кисть, сжимая большой палец, то его кончик выглядывает из кулака, длина среднего пальца превышает 10 см, а соотношение длины кисти в см и роста в метрах больше 11%, большим пальцем и мизинцем можно легко охватить запястье.
Данные инструментальных методов:
- ЭКГ – аритмия, гипертрофия сердечной мышцы.
- ЭхоКГ – расширение аорты, нарушения строения клапанов, повреждения хорд, увеличенный левый желудочек, видоизмененный митральный клапан.
- Рентген – аорта в восходящей части и сердце увеличены в размерах.
- КТ и МРТ подтверждают отклонения в строении магистральных сосудов и сердечных клапанов или стенок.
- Аортография выявляет признаки аневризмы и расслоения аортальных оболочек.
Показано обследование офтальмолога с биомикроскопией тканей глазного яблока, травматолога для диагностики нарушений функции суставов и позвоночника.


Подтверждение генетических мутаций проводится при помощи анализа ДНК. Типичным признаком синдрома Марфана является значительное превышение нормы выделения хондроитинсульфата и гиалуроновой кислоты с мочой.
Лечение синдрома Марфана
Терапия зависит от наличия и степени выраженности симптомов. В основном используют курсы профилактического медикаментозного лечения бета-блокаторами, витаминами и антиоксидантами для предотвращения прогрессирования сердечно-сосудистых патологических изменений. Рекомендуется санаторно-курортное лечение – ванны, массаж, электросон, магнитотерапия на суставы.
Показания к хирургическим вмешательствам:
- недостаточность смыкания створок клапанов, значительный обратный ток крови;
- расширение аорты свыше 5 см;
- мешковидные аортальные аневризмы больших размеров или симптомы расслоения стенки.
Если пациентка с синдромом Марфана беременна, то при родах используют кесарево сечение, при наличии выраженной декомпенсации может быть показано прерывание беременности.
В послеоперационном периоде для профилактики эндокардита и тромбоэмболии назначается курс антибиотиков и антикоагулянтов.
Для нормализации зрения проводится лечение оперативным или лазерным методом, хрусталик при смещении удаляется и меняется на искусственный, подбирают линзы или очки.
Если имеются значительные деформации скелета, то используют протезирование суставов, пластику грудной клетки, стабилизацию поясничного или шейного отдела позвоночника.
Прогноз для больных
Чаще всего продолжительность жизни зависит от степени сердечной недостаточности или угрозы разрыва аневризмы аорты. Проведение оперативного лечения в оптимальные сроки помогает пациентам сохранить здоровье до 60 — 70 лет. Если нарушения затрагивают несколько систем организма, то из-за различных осложнений 90% больных умирают молодыми.

Правила жизни
Больные с синдромом Марфана должны соблюдать такие рекомендации:
- Низкий уровень физических нагрузок, запрет на соревнования, профессиональный спорт, а особенно борьбу, подводное плавание, тяжелую атлетику.
- Нельзя поднимать тяжести более 3 кг.
- Избегать вредных условий производственной деятельности, противопоказана работа, требующая нагрузку на зрение, тяжелый физический труд.
- Запрещено пребывание в условиях высокой радиации или температуры.
Для детей физкультуру можно проводить только в специальных группах в виде лечебной гимнастики.
Наследование синдрома Марфана в наши дни
Врожденный синдром Марфана передается по аутосомно–доминантному типу. Это значит, что его появление в семье подчиняется таким закономерностям:
- чаще болен один из родителей;
- частота появления у девочек и мальчиков одинаковая;
- вероятность возникновения у ребенка равна 50%;
- от здоровых родителей (при наличии больных бабушек, дедушек) обычно не передается;
- если провести анализ родословного древа, то выявляется вертикальный способ передачи синдрома в каждом поколении;
- не все члены семьи имеют одинаковые признаки болезни.
Следует учитывать, что в 20% случаев мутации гена не связаны с наследственностью, они могут возникать первично. В таких случаях играет роль возраст родителей.
Доказано, что чаще дети с этой аномалией развития появляются, если отец старше 35 лет. В следующем поколении больных при таком варианте наследования может и не быть (пропуск поколения), но половина их потомков окажется с этой патологией.
Поэтому всем будущим родителям, у которых в семьях были выявлены лица с синдромом Марфана, нужно перед планированием беременности обязательно пройти консультацию в медико-генетическом центре.
Рекомендуем прочитать статью о внезапной коронарной смерти. Из нее вы узнаете о причинах внезапной остановки сердца, факторах риска, оказании первой помощи, а также мерах профилактики.
А здесь подробнее об узелковом периартерите.
Синдром Марфана связан с дефектом генов, обеспечивающих прочность соединительной ткани. Основную опасность представляет аномальное строение сердца и аорты. Причиной смерти чаще всего бывает расслоение аневризмы аортальной стенки.
-
Читайте также:

Кроме этого, особое строение костей скелета и нарушение зрения приводят к трудностям в социальной сфере. Диагностика в типичных случаях основана на внешних признаках, а для ее подтверждения используют инструментальные методы. Специфического лечения и профилактики болезни нет.